In brief¶
Microbial communities drive the cycling of energy, biomass, and nutrients on our planet, and are essential for the health of humans, animals, and plants. Taxonomic profiling of these communities, i.e. quantifying which taxa are present at what abundance, is a key step to, e.g., identifying the drivers of microbial community structure, discovering microbial disease biomarkers, and unraveling the complexity of microbial interactions.
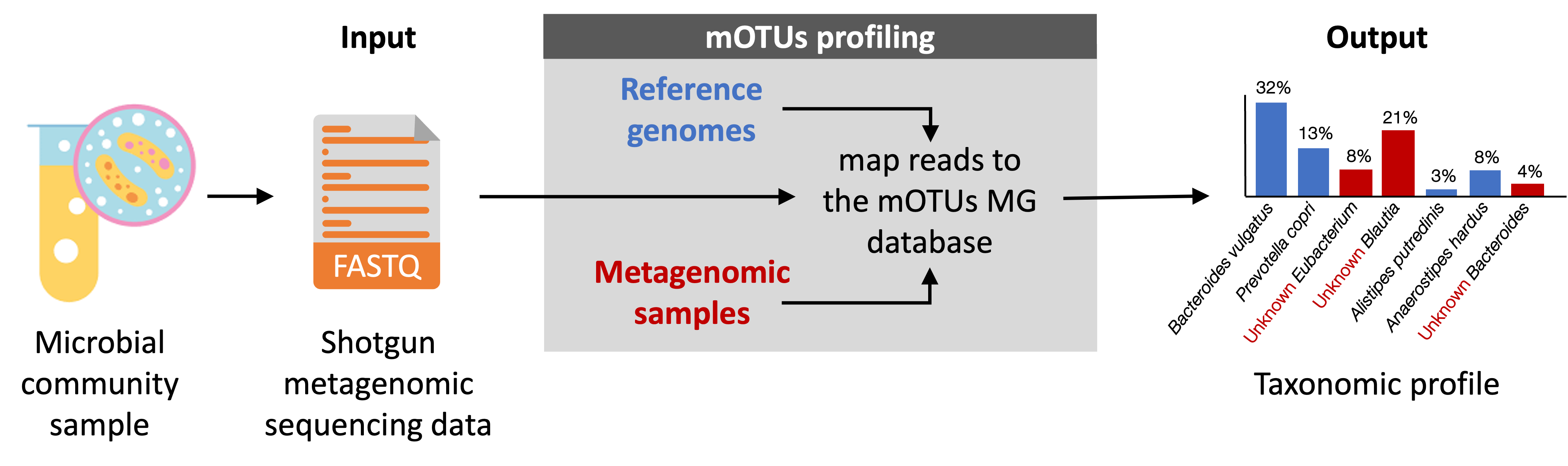
To enable the profiling of species-level composition of microbial community samples, we have developed mOTUs: a command-line tool for taxonomic profiling of shotgun metagenomic sequencing data. By using a set of universal single-copy marker genes (MGs), mOTUs is supported by, yet independent from, the availability of sequenced genomes from isolated strains. As such, it enables reference genome-independent profiling of both known and unknown species. The tool uses one or several FASTQ-formatted metagenomic sequence file(s) as input and outputs a taxonomic profile that reports the relative abundance of detected species either as read counts or proportions.
Statistics¶

Why should you use mOTUs?¶
It profiles over 124K species-level taxonomic units
The mOTUs database is constructed from 919K isolate and single cell-amplified (SAGs) genomes and 2.83M metagenome-assembled genomes (MAGs) generated from over 117K metagenomic samples spanning diverse microbiomes, which include (in addition to the human and ocean microbiome) soil, freshwater and gastrointestinal tract microbiomes of ruminants and other animals, environments we found to be greatly underrepresented by reference genomes.
In the current version, 124,295 species-level taxonomic units (mOTUs) were constructed using sequences of 10 single-copy marker genes recovered from these genomes. 30,256 mOTUs are represented by an isolate genome, whereas 94,039 mOTUs are represented by MAGs only.
It identifies and quantifies species with high precision
Due to the compositional nature of metagenomic data, methods that rely on quantifying only species for which reference genomes are available produce biased results in which the relative abundance of such species in a microbial community is overestimated. In addition to profiling both known (depicted in blue in the example below) and unknown (red) species represented in its MG database, mOTUs estimates the fraction of microbes which are not represented in its database but in the input metagenome, i.e the unassigned fraction (black).

Furthermore, mOTUs focuses on high precision, i.e. on detecting as many correct taxa (true positives) as possible while minimizing the detection of wrong ones (false positives). Together, these steps ensure a high quality of taxonomic profiles generated with mOTUs, which has been corroborated in independent benchmarks such as LEMMI and CAMI.
It provides the user with genomic context for all profiled species
You can browse and query all 3.7 million genomes used for the construction of the mOTUs MG database at https://motus-db.org/. The database allows you to search for mOTUs by taxonomy or genomes by quality scores, for example. You can also download selected genomes that represent a taxonomic unit of interest (up to 200), or use the mOTUs download tool for more extensive downloads.
It has an extendable database
In addition to the over 3.7 million genomes included in the mOTUs database, you can use the mOTUs extender tool to add genomes and profile additional taxa of interest.
It can profile long-read sequencing data
Long-read sequencing is becoming more and more popular for sequencing DNA from microbial communities or isolated strains.
There are few tools available for taxonomic profiling of long-read sequencing data, most of which rely on classifying and counting reads.
The long-read profiling pipeline in mOTUs performs reasonably well when compared with other pipelines specifically developed for long-read
sequencing data (see Portik et al., 2022).
You only need to run motus prep_long -i input_file -o output_file before running the regular profiling workflow.
It is easy to install, easy to use, and resource efficient
You can easily install mOTUs using conda or pip. Once installed, you can obtain a taxonomic profile with a single shell command. To increase accessibility, mOTUs is now also available as a QIIME2 plugin .
mOTUs is faster and requires less memory than most taxonomic profiling tools. The figure below shows data from the independent benchmark study LEMMI.


![]() mOTUs is part of the ELIXIR-CH Service Delivery Plan.
mOTUs is part of the ELIXIR-CH Service Delivery Plan.